![]()
Diplôme Universitaire de
Bioinformatique
Structurale
Olivier Alméras

Laboratoire de Cancérologie Expérimentale
Département des Sciences du Vivant
Dirigé par Sylvie CHEVILLARD
Sous la responsabilité de Nicolas UGOLIN
Analyse des données de puces à ADN
Les projets de séquençage permettent de déterminer la
composition du génome de nombreuses espèces. Toutefois le nombre
et l'organisation des gènes au sein des génomes restent à déterminer.
Les techniques informatiques pour reconnaître les unités de transcription
dans l'information génomique brute sont en cours de développement.
Dans le cas du génome humain, les estimations du nombre de gènes
sont comprises entre 26 000 et 100 000 gènes (IHGSC, 2001: 30 000-40
000 gènes, Venter et al., 2001: 26 000-38 000 gènes). Au regard
de ces avancées pour connaître le génome, le défi
reste de déterminer le rôle de tous ces gènes en terme
d'expression, de régulation et de fonctionnement physiologique de l'organisme.
Notamment la génomique fonctionnelle s'intéresse à l'expression
et à la régulation du génome. Des questions essentielles
peuvent alors se poser telles que :
Où sont localisées les expressions de tous ces gènes ?
Comment se modifie l'expression du génome suite à un changement
physiologique ou un stress extérieur ?
Durant des années, les biologistes moléculaires ont focalisé les
recherches sur un seul gène ou quelques gènes à la
fois. Les techniques se basaient sur l'utilisation de sondes d'acides
nucléiques
(hybridation in situ, Northern blotting,...) ou d'anticorps (immunocytochimie,
Western blotting,...). Les descriptions d'expression de gène dépendaient
de la disponibilité de ces sondes et de l'intuition des chercheurs.
L'information sur le génome et, en parallèle, le développement
des méthodes SAGE (Velculescu, 1997) et des techniques des puces à ADN
(microarray) ont fourni les moyens pour réaliser une analyse vectorielle
de l'expression de milliers de gènes en une seule expérience.
Les résultats donnent une estimation des niveaux d'expression
des gènes
inclus sur la puce à ADN dans un type cellulaire, un tissu ou
un organe particulier. Le concept de base des analyses sur puce à ADN
est simple (Fig.1). L'ARN est
collecté à partir d'un type
cellulaire ou d'un tissu d'intérêt. Son marquage génère
l'ensemble des cibles d'acides nucléiques libres dont l'identité ou
l'abondance est recherchée. Ces cibles sont ensuite hybridées
aux séquences
de sondes d'ADN correspondant aux gènes spécifiques, fixés à un
support solide suivant une configuration connue. L'hybridation, basée
sur l'appariement des bases de Watson et Crick, entre la sonde et la
cible, fournit une mesure quantitative de l'abondance d'une séquence
particulière
dans la population cible. Cette information est enregistrée numériquement,
et soumise à différentes analyses pour en extraire l'information
biologique. La comparaison des profils d'hybridation permet l'identification
des ARNm qui diffèrent en abondance dans deux échantillons
ou plus. Ainsi les puces à ADN fournissent un outil puissant pour
cribler des échantillons biologiques sur les modifications de
l'expression des ARNm, qui accompagnent et peuvent réguler un
changement physiologique ou pathologique.
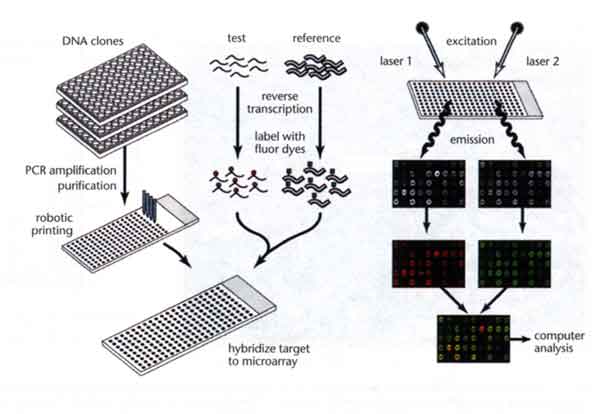 |
Fig.1
Schéma d'une étude par puce à ADN
|
Avant d'aborder
les limitations de la technique, intéressons nous aux raisons de l'engouement
qu'elle a suscité au cours des dernières années. Ainsi
les techniques de puces à ADN trouvent leur utilité dans plusieurs
champs d'applications, tels que:
- La découverte de gène: description des voies de régulation,
basée sur l'hypothèse que des gènes régulés
en parallèle partagent des mécanismes de contrôle communs.
- Le diagnostique: identification de profils d'expression de gènes qui
caractérisent des états pathologiques et qui peuvent représenter
des indicateurs de pronostiques.
- La découverte de médicaments et toxicologie...etc.
Toutefois l'apparente simplicité du concept des puces à ADN masque
des problèmes considérables. Et toutes les promesses de la technique
ne sont pas encore tenues. Les puces à ADN sont très récentes:
les méthodologies sont en cours d'évolution, des standards communs
sont encore à établir et de nombreux problèmes subsistent
avec les schémas expérimentaux, et la variabilité des
données doit être mieux comprise et surmontée. Chaque étape
dans le déroulement de l'expérience peut être source d'erreurs.
Dans l'étude qui suit, sera présenté un exemple de protocole suivi pour générer les données, de l'extraction du matériel génétique à l'obtention des rapports d'expression entre un échantillon et son témoin. Puis nous décrirons les logiciels utilisés pour l'interprétation des données. En particulier, nous présenterons l'approche originale pour réduire l'influence du bruit de fond par l'application de filtre lors du traitement de l'image, la résolution du problème du positionnement des grilles de lecture des signaux et la méthode pour associer à un gène une probabilité d'expression. Enfin nous aborderons les deux programmes développés lors de cette étude, pour le repositionnement manuelle des grilles de lecture et l'estimation des probabilités d'expression d'un gène.
A.
Protocole expérimental
Le TIGR propose un protocole standard publié par Hedge et al. (2000).
Le descriptif de la préparation des puces à ADN qui suit, est
principalement basé sur cette publication de référence.
1. Fabrication des lames
Les conditions de préparation des puces à ADN induisent de fortes
variations dans la morphologie des spots obtenus, et dans la quantité de
matériel génétique retenue sur la lame. Ainsi la préparation
des logiciels d'analyse des puces à ADN doivent tenir compte des
méthodes
employées dans l'obtention des puces. Un spot
homogène ne sera
pas analysé de la même manière qu'un spot avec de très
fortes concentrations d'ADN fixées en périphérie du
spot. Dans un spot hétérogène, l'analyse du signal
devra être
effectuée au niveau de chaque pixel, tandis qu'un spot homogène
pourra être analysé par une valeur médiane sur l'ensemble
du spot.
Une puce à ADN est constituée de clones d'ADNc ou de gènes
amplifiés par PCR, et fixés sur une lame de microscope en verre
dérivatisé (Hegde et al., 2000). Pour l'analyse de l'expression
chez les eucaryotes, les données des marqueurs de séquences exprimées
(EST) représentent les données les plus fournis pour l'identification
de gènes. Elles correspondent à une portion d'une séquence
d'ADNc. L'objectif des banques d'ADNc est de regrouper dans une collection
l'ensemble des gènes susceptibles de s'exprimer. Par exemple, le TIGR
Human Gene Index contient 48000 clones d'ADNc . La banque de gènes retient
en priorité des gènes connus avec une information cartographique
(position sur le chromosome). Un certain nombre de clones sont également
représentatifs de gènes avec une fonction inconnue. La banque
UniGene au NCBI est basée
sur une classification d'EST, tandis que le TIGR tente d'assembler les EST d'une classe, produisant des tentatives de séquences
consensus humaines (THC).
a) Amplification
PCR
La PCR permet de disposer d'une grande quantité de matériel génétique à partir
de 1 µl de culture cellulaire, provenant des banques de gènes
ou des sondes spécifiques développées au laboratoire.
Les clones sont multipliés par culture durant une nuit. Diluées
dans l'eau, les cellules sont éclatées. L'ADN est amplifié par
plusieurs cycles de réplication. Les produits de PCR sont purifiés
sur des plaques 96 puits à filtre en verre. Ils sont fixés dans
une forte concentration saline (1:5 Guanidine-HCl 5,3M/KAc 150 µM). Une élution
dans un mélange eau-tampon TE termine la purification.
Pour 30000 clones, Hegde et al. obtiennent des taux d'amplification d'une
simple bande de 87,5%, de 6,3% pour les bandes faibles ou multiples et 6,2%
sans amplification
(Fig. 2). Au laboratoire, les concentrations des produits de PCR sont comprises
entre 20 et 100 ng/µl.
 |
Fig.2
Produits de PCR de 96 clones d'ADNc
directement à partir de culture de bactérie La ligne de gauche à chaque bande du gel contient un marqueur de taille de 1kb. Chaque bande contient 24 échantillons distinctes. |
b) Fixation des sondes sur les lames
Les produits de PCR resuspendus dans un tampon dénaturant sont déposés
sur les lames de verre coatées de poly-L-Lysine ou d'aminosilane grâce à un
robot (Fig. 3). 5 µl de produits PCR resuspendus permettent de préparer
au moins 100 lames. Chaque spot reçoit un dépôt d'environ
2 ng d'ADNc. Toutefois quelques paramètres, décrits ci-dessous,
influencent la quantité effectivement retenue sur la lame. Par exemple,
le robot d'Intelligent Automation Systems dépose des spots de 130 µm
de diamètre moyen sur des lames couvertes de silane. 19200 unités
d'hybridation peuvent être fixés sur une seule lame.
 Fig.3
Robot préparateur de lame
Fig.3
Robot préparateur de lame
La surface de la lame et le tampon de dépôt sont les deux composants
critiques pour la reproductibilité et la fiabilité des analyses
de puces à ADN. La plupart des publications utilisent un tampon de 3xSSC
et des lames coatées de poly-L-Lysine. Toutefois Hegde et al. suggère
que l'aminosilane offre une surface plus fiable avec deux fois moins de bruit
de fond de fluorescence comparé à la poly-L-Lysine, pour une
intensité de signal significativement supérieure.
Le tampon 3xSSC peut avantageusement être remplacé par le DMSO
50% pour les lames d'aminosilane. Il donne les intensités d'hybridation
les plus élevés, fournissant plus d'ADN simple brin et une meilleure
fixation (Fig. 4).
 |
Fig.4
Effets de différents tampons de spotting et de protocoles
de purification de l'ADN sur la
fixation de l'ADN et l'hybridation sur lame coatée d'aminosilane CMT-GAPS Cette image en couleurs artificielles a été générée par le dépôt d'échantillons identiques dans les lignes adjacentes. Une même sonde d'ARNm marquée est hybridée sur tous les spots. Les lignes rouges séparent les paires de lignes. Les paires de lignes 1-3 et 5-7 contiennent les échantillons contenant soit le tampon DMSO 50%, soit le 3xSSC. En comparant les spots adjacents verticalement, il apparaît que le DMSO permet une hybridation avec des intensités d'hybridation d'au moins 1,5 fois supérieures à celle du SSC. Les paires de lignes 4 et 8 montrent les effets de différents protocoles de purification sur la fixation de l'ADN et l'hybridation. La méthode de filtre de verre (Glass Filter) donne les meilleurs résultats comparés à la précipitation à l'éthanol. |
Le mode opératoire pour déposer l'ADN sur les lames influence la morphologie des spots. Les trois types suivants peuvent être distingués : dépôt par électropolymérisation, dépôt sans contact, qui produit des spots homogènes, et le dépôt avec contact, qui donne des spots hétérogènes. La méthode décrite par Hedge et al. (2000) utilisent la capillarité pour charger les aiguilles et les interactions de la tension de surface pour libérer la solution sur la lame. Les paramètres tels que les accélérations du bras du robot, la température et l'humidité contrôle la morphologie, la taille et la position des spots. Selon Hedge et al., l'humidité relative de 45% et une température de 22°C sont optimum (Fig. 5). Des changements dans l'humidité et la température modifient la taille et la morphologie des spots, ainsi que l'efficacité de la fixation de l'ADN sur la lame.
 |
Fig.5
Effet de la température et de l'humidité sur
la morphologie des lames
Le première série de spots commence avec une température de 22,2°C et une humidité relative de 45-50%, qui ont été déterminées comme étant optimum. Sous ces conditions, les spots ont une apparence homogène. Ensuite, la température est réduite à 16,7°C, entraînant une réduction des spots, qui sont moins distincts. La température et l'humidité ont ensuite été augmentés. Les lignes de spots larges dans le milieu de la lame (lignes 13-15) ont été obtenues à 26,7°C et 80-85% d'humidité relative. Lorsque la température et l'humidité diminuent à nouveau, on repasse par les conditions optimums. Les clones d'ADNc ont été hybridés avec une sonde spécifique du vecteur, marquée au Cy3. |
Protocole simplifié:
1.Ajout volume à volume des produits de PCR au DMSO dans une plaque
96 puits.
2.Dépôt des produits de PCR à 22°C et 45% d'humidité relative.
Eviter poussières et traces de doigts.
3.Séchage et fixation de l'ADN par un UV-crosslinking à 90-350
mJ.
4.Les lames imprimées sont stockées à l'abris de la lumière
et de l'humidité, à température ambiante.
2. Préparation des échantillons
Comme de nombreuses expériences basées sur l'ARN, la pureté et
la qualité des ARN de départ a un effet significatif sur les
résultats de l'expérience. En effet, les ARN sont rapidement
dégradés par des ribonucléases dans les cellules. Ce mécanisme
de régulation complexe permet un contrôle de la quantité des
ARN, et ainsi de l'expression des gènes (un exemple de contrôle
de la dégradation de l'ARNm du TNF alpha est décrit par Stoecklin
et al., 2003).
a) Extraction des ARN
Les impuretés des préparations d'ARN ont des effets négatifs
sur l'efficacité du marquage et la stabilité des cibles fluorescentes
utilisées sur les puces à ADN. Hedge et al. (2000) considère
que le Trizol (Life Technologies) atteint une qualité reproductible
satisfaisante des ARN à partir de culture cellulaire ou de plusieurs
types de tissus. Une étape supplémentaire est nécessaire
pour éliminer les polysaccharides lorsque les ARN proviennent de
tissus.
Protocole simplifié:
1.Reprendre les cellules avec un tampon PBS, en éliminant le milieu
de culture.
2.Ajouter le Trizol et disrupter le culot par plusieurs passages dans
une seringue.
3.Incuber 5 mn à température ambiante.
4.Compléter la lyse cellulaire par un mélange Chloroforme:Trizol
(1:5).
5.Eliminer les débris cellulaires par centrifugation à 4°C.
6.Précipiter l'ARN à l'isopropanol.
7.Centrifuger, et resuspendre l'ARN dans l'ethanol 70%. L'ARN peut être
stocké dans l'éthanol à 70% à -20°C jusqu'à son
utilisation.
8.Avant le marquage, centrifuger et resuspendre le culot dans un tampon
exempt de RNase.
b) Marquage des sondes d'ADNc
La quantité d'un échantillon prélevé sur un patient
est souvent très limitée. Les protocoles fonctionnant avec peu
d'échantillon de départ ont été privilégiés
par Hedge et al. (2000). Les cibles marquées s'obtiennent en intégrant
des déoxyribonucléotides marqués lors de la synthèse
du premier brin de l'ADNc à partir d'ARN. Les ARN totaux s'avèrent
aussi efficaces que les ARN poly(A+) avec 20 fois moins de matériel
de départ (Fig. 6).
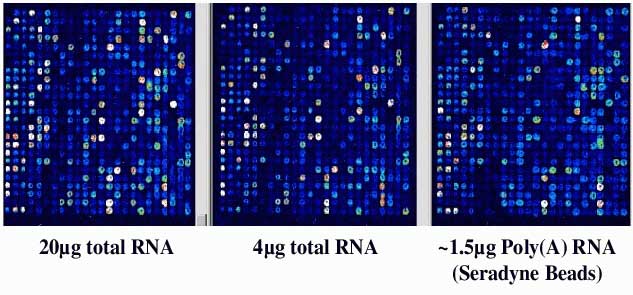
Fig.6
Comparaison des cibles d'hybridation faites à partir
d'ARN total et d'ARN poly(A+)
Les blocs correspondants de puces à ADNc contenant 7200 éléments
sont hybridées chacun avec une cible
marquée préparée à partir soit de 20 µg d'ARN
total, soit de 4 µg d'ARN total, soit de 1,5 µg d'ARN sélectionnés
par
leur queue poly(A+). Les intensités d'hybridation relative sont similaires
pour chacun des échantillons d'ARN total, et
elles sont légèrement plus élevées que celles des
ARN poly(A+). Toutes les images sont obtenues avec le même
laser et des paramètres PMT durant le scannage. L'affichage utilisent
les mêmes paramètres.
La synthèse du premier brin d'ADNc intègre des nucléotides
marqués par des amino-allyl. L'encombrement stérique réduit
de cette modification permet une grande homogénéité de
marquage. Les fluorochromes sous forme NHS-ester (SE, succinimidyl ester) sont
ensuite liés de manière covalente (Fig. 7).
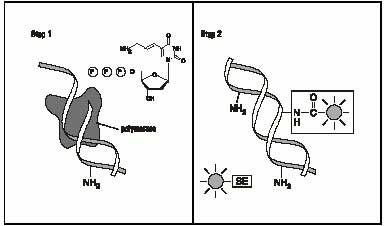 |
Fig.7
Diagramme schématique de la liaison du
fluorochrome-SE sur l'amino-allyl du nucléotide. 1- Les dUTP-amino-allyl sont incorporés enzymatiquement. 2- Un fluorochrome activé est utilisé pour marquer le radical amino-allyl incorporé. |
Protocole simplifié:
1.Le mélange de marquage contient un rapport 2:3 de dUTP-aminoallyl:dTTP
non marqué avec une concentration équivalente de chacun des autres
nucléotides (dATP, dCTP, dGTP).
2.À 10 µg d'ARN total, hybrider des amorces hexamers aléatoires.
3.Aux ARN totaux, ajouter la reverse transcriptase et le mélange de
nucléotides.
4.Incuber au moins 3h à 42°C.
5.Hydrolyser les ARN avec NaOH 1M et EDTA 0.5M, à 65°C.
6.L'hydrolyse est neutralisée par du HCl 1M.
L'ADNc-aa est séparé des dUTP-aa non incorporés et des
amines libres avant la conjugaison au fluorochrome, SE. La purification est
effectuée sur colonne de purification des produits PCR de Qiagen (QIAquick)
ou par centrifugation sur Microcon 30.
Les deux échantillons, dont les niveaux d'expression des ARN vont être
comparés, sont marqués par deux fluorochromes de longueurs d'ondes
différentes. Les plus classiques sont les cyanines Cy3 et Cy5.
Protocole simplifié:
1.Resuspendre l'ADNc-aminoallyl dans un tampon carbonate.
2.Ajouter le fluorochrome-SE repris dans du DMSO anhydre.
3.Incuber 1h à l'abris de la lumière à température
ambiante.
Après le couplage covalent du fluorochrome, les ADNc-aminoallyl non
couplés sont éliminés avec le kit de purification QIAquick.
3. Hybridation sur les lames
Le but est d'obtenir une forte spécificité avec un minimum de
bruit de fond.
- Choix du nombre de répétitions
Habituellement pour surmonter les erreurs d'estimation d'une valeur, une
expérience
prévoit de répéter les mesures afin de déterminer
sa valeur moyenne et la variance associée. Avec les puces à ADN,
la multiplication des lames est coûteuse, longue et parfois limitée
par la quantité de matériel génétique disponible.
De plus, la simple répétition du dépôt des sondes
permet seulement de corriger les mesures des erreurs du robot et de l'hétérogénéité de
la lame.
Pour éviter le biais lié à une incorporation préférentielle
en fonction de la séquence des fluorochromes, chaque expérience
et son témoin sont réalisés en double en inversant les
fluorochromes. Cette technique est appelée «flip-flop» au
laboratoire (Reverse labeling ou label switching, Kerr et Churchill, 2001 et
Ugolin, 2002). Deux lames sont donc préparées avec les mêmes
sondes. Les cibles de l'échantillon traité, marqué au
Cy3, et l'échantillon non traité (le témoin), marqué au
Cy5, sont déposées sur la première lame. Pour la deuxième
lame, les fluorochromes sont inversés entre l'échantillon traité et
son témoin.
De plus, un gène peut être représenté plusieurs
fois sous forme de séquences différentes. Ceci résulte
soit de la redondance du gène lors de la préparation des banques
d'ADNc (souvent due à l'abondance d'un ARN particulier), soit d'un choix
délibéré de répéter un gène d'intérêt.
Ainsi le risque de passer à côté de l'expression de ce
gène d'intérêt est réduit par l'hybridation sur
des séquences différentes.
Environ la moitié des gènes est représentée par
un seul spot. 20% des gènes ont deux spots. 10% des gènes ont
trois spots, etc... Le plus représenté dans une étude
sur le cerveau humain au laboratoire va jusqu'à 14 spots pour un même
gène.
- Type de sondes
La puce à ADN contient des contrôles négatifs contenant
du tampon seul et des séquences de levure, qui ne reconnaissent pas
les cibles d'échantillon humain. Les contrôles positifs sont préparés
avec des concentrations croissantes d'ADNc de gènes de ménage.
Ces gènes s'expriment en permanence pour entretenir un bon fonctionnement
de la cellule. Les échantillons contiennent ainsi une concentration
relativement homogène d'ARNm de gènes de ménage.
Les ADNc proviennent essentiellement de banque de gènes. Tous les fragments
sont insérés dans le même vecteur. Les amorces de PCR pour
préparer les sondes sont identiques, et seule la taille de l'insert
varie. Ainsi les PCR sont réalisées dans les mêmes conditions,
et les résultats d'amplification sont très homogènes.
Cette homogénéité n'est pas retrouvée avec les
sondes préparées à partir d'oligonucléotides spécifiques.
Les conditions de la PCR sont pourtant les mêmes que pour les ADNc issus
de la banque, mais les amorces de PCR varient en fonction de la nature de la
séquence de l'oligonucléotide.
b) Mélange des sondes et fixation sur la lame
L'ADN est ensuite déposé sur un support, principalement en verre
en raison de son bon compromis entre le coût et ses propriétés
optiques avantageuses (Diehl 2001). La surface du verre est activée
avec différents groupements fonctionnels: aminosilane, poly-L-Lysine...
De la qualité de la fixation dépendront la limite de sensibilité et
l'intervalle dynamiques des mesures. La quantité et l'homogénéité de
l'ADN sur la lame améliorent l'analyse d'image, et augmentent la reproductibilité de
la détection du signal. Hedge et al. (2000) considère les lames
d'aminosilane comme les plus efficaces. Avant l'hybridation, les groupements
fonctionnels d'amine libre sur la lame doivent être bloqués, sinon
une fixation non spécifique des cibles marquées d'ADNc produira
du bruit de fond et réduira la quantité de cibles disponibles.
Une préhybridation avec une solution contenant de l'albumine bovine à 1%
est très efficace pour éliminer les fixations non spécifiques.
Immédiatement après la préhybridation, les cibles sont
hybridées suivant le protocole suivant:
1.Chaque cible est reprise dans 12 µl de tampon d'hybridation.
2.Les 12 µl des cibles marquées au Cy3 et au Cy5 sont combinés,
et vigoureusement mélangés. Les fixations non spécifiques
sont bloquées par l'ajout d'ADN-COT1 et d'ADN-poly(A).
3.En chauffant à 95°C pendant 3 mn, les doubles brins des cibles
sont dénaturés en simples brins.
4.Centrifuger, et appliquer les cibles marquées sur la lame préhybridée.
5.Incuber pendant 16-20 h à 42°C dans une chambre d'hybridation
scellée.
6.Rincer, et laisser sécher dans une atmosphère sèche.
(Fig.8)
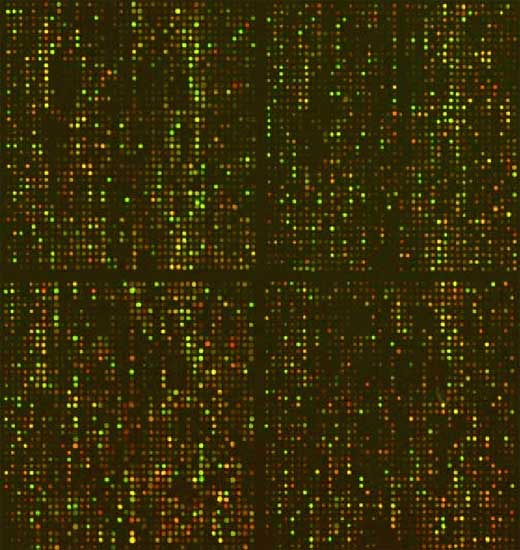 |
Fig.8
Hybridation d'ARNm sur une partie d'une puce à ADN de 19200 éléments.
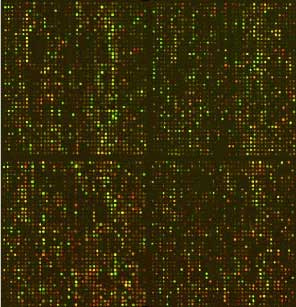
10 µg d'ARN extrait d'un carcinome de colon humain KM12L4A (expérience) et d'une lignée de référence KM12C(témoin) sont utilisés pour une transcription inverse, puis les ADNc sont marqués au Cy5 et Cy3 respectivement. Ces cibles sont ensuite hybridées sur la puce à ADN portant 19200 sondes distinctes de clones d'ADNc humains. Le contraste de l'image a été ajusté pour permettre une visualisation satisfaisante sur l'ensemble des gènes. |
4. Acquisition
des images par scanner
Les deux grandes catégories de scanner se partagent entre les scanners
non-confocaux et les scanners confocaux. Les non-confocaux offrent un angle
de capture large, mais ils font une moyenne des signaux. Les signaux parasites
se retrouvent mélangés avec le signal convenable. Grâce à la
grande précision de la zone excitée sur le scanner confocal,
la fluorescence faible peut être mesurée à côté de
zone à forte fluorescence (Dixon, 2001). Toutefois les indices de réfraction à deux
longueurs comme celles du Cy3 et du Cy5 sont différentes, et les surfaces
de capture ne sont pas équivalentes. Ainsi la superposition des images
n'est pas possible. Une exception à cette règle, Brown et al.
(2001) ont développé un scanner spécial pour que les images
soient superposables aux deux longueurs d'onde. Ainsi l'analyse d'image doit
tenir compte de la manière dont l'image a été scannée.
Dans le laboratoire, l'expression différentielle de gènes s'obtient
en scannant les puces à ADN hybridées à l'aide d'un
scanner à laser
confocal. Ce laser excite à leur longueur d'onde les fluorochromes
vert (Cy3) et rouge (Cy5), respectivement 543 nm et 633 nm. Le Cy5 peut être
scanné en premier, car il est plus sensible à la photodégradation
que le Cy3. L'émission de fluorescence est lue avec le filtre d'émission du
Cy3 dans le canal 1, et celui du Cy5 dans le canal 2. Pour chaque fluorochrome,
le scanner
produit deux images TIFF séparées, en format 16 bit (65535
niveaux de gris). Ces images sont analysées pour calculer les niveaux
d'expression relative de chaque gène, et identifier les expressions
différentielles de gènes.
B. Traitement des données brutes
L'analyse des puces à ADN souffre de biais intrinsèques à la
méthode. Nous avons donc cherché à apporter des réponses à ces
problèmes par une délimitation automatique des spots, une segmentation
poussée des pixels de chaque spot en prenant en compte à la fois
le bruit de fond intra- et extra-spot et les signaux parasites. La suite logicielle
permet également une correction des signaux de fluorescence en utilisant
de façon originale le dye-swap, la détermination d'un facteur
de reproductibilité (utilisable pour réaliser du clustering flou),
de même que la normalisation des rapports d'induction ou de répression
par recherche automatique et systématique de la population invariante.
1. Identification des spots
Dans un premier temps, les spots représentant les gènes doivent être
repérés et distingués des artefacts de l'hybridation,
dus par exemple à la précipitation de cibles ou à des
contaminants (poussières,...) sur la surface de la lame. Le repérage
de la position des spots s'appuie sur l'arrangement régulier des dépôts
automatisés. Toutefois les intensités variables des signaux et
le bruit de fond s'ajoutent aux irrégularités possibles de la
grille de dépôt pour nécessiter un contrôle. Généralement,
la localisation des spots est couplée avec l'analyse du bruit de fond.
Pour les puces à ADN, il est important de calculer le bruit de fond
spécifiquement à chaque spot, plutôt que globalement pour
l'ensemble de la lame ou uniquement autour du spot.
2. Détermination du bruit de fond
a) Nature du bruit de fond
La faible reproductibilité des analyses sur puce à ADN a été attribuée à un
ensemble de facteurs comprenant les conditions de collecte des cellules, la
variation biologique, la qualité de l'ARNm, la qualité du dépôt
des spots, l'hybridation et l'incorporation différentielle des nucléotides
fluorescents. Martinez et al. (2003) ont identifié une fluorescence
contaminante, localisée au niveau du spot, dans le canal du Cy3 sur
plusieurs lames commerciales et sur les puces fabriquées dans leur laboratoire.
Un scan avant l'hybridation ne peut pas prédire la contribution de cette
fluorescence contaminante après hybridation. Afin cette dernière
est moins observée avec le fluorochrome Cy5. La plus faible énergie
du Cy5 par rapport au Cy3, nécessaire à son excitation, pourrait
expliquer cette minimisation. En plus de ce bruit de fond contaminant dans
le spot, très variable d'un spot à l'autre, il existe un bruit
de fond relativement homogène sur l'ensemble de la lame de verre.
b) Estimation du bruit de fond par segmentation du signal
Généralement, l'estimation du bruit de fond est locale (autour
du spot) ou globale (zone de référence sur la lame).
Mais la nature et l'intensité du bruit de fond en dehors du
spot (local ou global) sont différentes de celles à l'intérieur
du spot. Habituellement les chercheurs utilisent un rapport (signal
- bruit de fond)/(écart-type
du bruit de fond) supérieur à un seuil de 3 ou plus
pour valider un signal. Toutefois le bruit de fond est calculé en
dehors du spot. Or le bruit de fond calculé dans le spot peut
ne pas être de même
valeur que celui en dehors du spot (Fig. 9). Martinez et al. (2003)
montrent que cette méthode valide des spots sur lesquels rien
n'est hybridé.
Ils proposent deux solutions: une exposition pendant 4h à l'air
avant le dépôt des sondes pour réduire l'affinité avec
les contaminants fluorescents, et l'utilisation d'algorithmes de
courbes de résolution multivariées permettant de prendre
en compte les contributions spectrales du signal Cy3, du verre et
de la fluorescence contaminante. Mais les scanners commerciaux ne
sont pas encore adaptés à cette dernière méthode.
De plus, ne connaissant pas la nature de la fluorescence des polymères
d'ADNc dans un mélange avec des contaminants fluorescents,
il est difficile d'utiliser les résultats de l'analyse de
courbes multivariées.
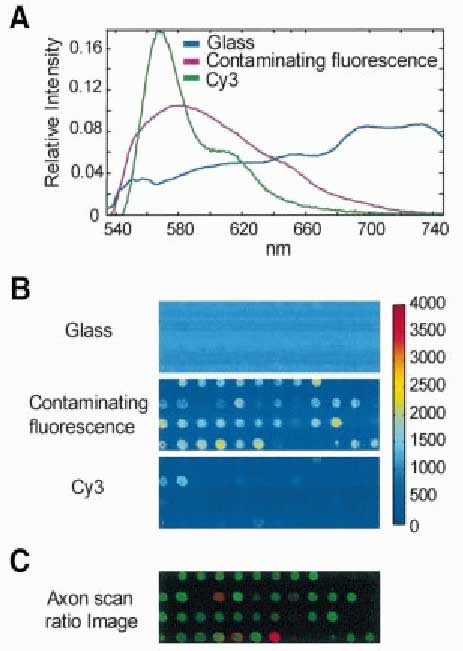 |
Fig.9
Résultats d'un scan hyperspectral
d'une lame Corning hybridée montrant la fluorescence contaminante en présence de Cy3. Les régions de la puce à ADN hybridées avec l'ADNc marqué au Cy3 et au Cy5 sont scannées avec des longueurs d'onde d'émission de 540 nm à 740 nm avec un scanner Axon 4000B et une résolution de 10 µm. Le spectre et les écarts de concentrations (concentration map) sont générés à partir de l'analyse d'image multivariée des images hyperspectrales contenant 46800 spectres sur une zone de 3,9 x 2,3 mm². Chaque image est calibrée proportionnellement à l'intensité totale des images combinées de l'Axon 4000B pour une comparaison visuelle. Les facteurs de calibration appropriés sont calculés en multipliant chaque spectre par son écart de concentration et en appliquant une fonction de filtre similaire au filtre optique utilisé sur le scanner commercial (Axon). (A)
Spectre d'émission des espèces fluorescentes, normalisé
à
l'unité de longueur. Ces émissions de fluorescence
seraient
toutes confondues sur les scanners commerciaux de puce à
ADN. |
Une autre approche consiste à améliorer l'homogénéité du signal d'hybridation et du bruit de fond dans le spot. La variance du bruit de fond peut être réduite en moyennant les données de plusieurs images. Mais la diminution du signal, lorsque les fluorochromes sont trop longtemps excités (photobleaching), limite le nombre de passages du scanner. Une alternative (Ugolin et al., brevet, 2002) consiste à faire une moyenne entre 4 pixels de l'image. De plus, un filtre est appliqué à cette étape pour éliminer les pixels les plus hétérogènes. L'intensité moyenne dans le spot reste pratiquement inchangée, avec une variance réduite. Par ailleurs, l'intensité du bruit de fond diminue en moyenne (les pixels allumés sont écartés des pixels globalement éteints en dehors du spot) et en variance.
Après ce premier filtre, les pixels d'un spot sont ordonnés afin de déterminer leur type: signal d'hybridation, bruit de fond et éventuellement signal contaminant dans le spot. Un algorithme EM (Estimation Maximisation) avec 2 ou 3 classes segmente les pixels en différentes populations. Il est basé sur une distribution gaussienne modifiée. La moyenne et la variance de chaque classe sont obtenues par converge du maximum de vraisemblance après plusieurs itérations d'estimation et de maximisation. Afin d'éluder les intensités intermédiaires entre le signal d'hybridation et le bruit de fond, la méthode est modifiée pour ajouter un seuil à la probabilité de chaque pixel d'appartenir à l'une des classes. La classe du signal d'hybridation contient généralement la population la plus importante de pixels. Si aucune classe ne se distingue de celle du bruit, le spot est écarté des études ultérieures.
Pour comparer les différents traitements de l'image, Ugolin et al. (2002) propose un facteur Q. Il quantifie l'intérêt apporté par un traitement particulier sur le mélange du signal d'hybridation et du bruit de fond. Ainsi le protocole de filtrage minimise le bruit de fond sans altération du signal d'hybridation. De plus, l'algorithme EM diminue la variance du bruit de fond et du signal d'hybridation. Par rapport aux méthodes basées sur le bruit de fond en dehors du spot, cette approche permet de conserver et d'évaluer d'avantage de spots.
3. Correction des intensités
Une fois les spots identifiés et le bruit de fond local déterminé,
les intensités d'hybridation pour chaque spot doivent être corrigées
en leurs soustrayant l'intensité du bruit du fond. Une mauvaise estimation
du bruit de fond inclus dans le spot crée des sources d'erreurs, en
particulier pour les faibles intensités.
Grâce à la segmentation par l'algorithme EM, la distribution des
pixels du signal d'hybridation est distinguée de celle des pixels du
bruit de fond dans le spot. Les maxima de ces deux distributions fournissent
l'estimation du signal d'hybridation bruité et du signal de la lame.
Comme ces signaux sont cumulés, le signal d'hybridation est obtenu en
soustrayant le signal de la lame au signal d'hybridation bruité.
4. Normalisation
au laboratoire
Suite au traitement de l'image, les intensités de fluorescence relatives
doivent être normalisées dans chacun des deux canaux de longueur
d'onde scannés. La normalisation est nécessaire pour ajuster
les sources de biais systématiques. Ces variations sont dues entre autres à des
différences de propriétés physiques des fluorochromes
(sensibilité à la chaleur et à la lumière, durée
de demi-vie), d'efficacité d'incorporation des fluorochromes, à des
différences de quantités d'ARN de départ pour les deux échantillons
examinés, ou encore aux paramètres de réglage du scanner
(Yang, 2002).
a) Hybridation différentielle (Reverse labelling)
La stratégie de normalisation du laboratoire est basée sur deux
lames d'hybridation différentielle. Le schéma expérimental
implique l'obtention de quatre jeux de données. Un échantillon
ayant été soumis à un traitement particulier est comparé à un
témoin non traité. Ces deux sources de cibles servent à réaliser
l'hybridation compétitive sur la puce à ADN. L'hybridation compétitive
est réalisée avec l'échantillon marqué au Cy3 contre
le témoin marqué au Cy5. Les données sont doublées
avec la même expérience en inversant les fluorochromes. Ce schéma
est cité dans plusieurs articles, dont Kerr et Churchill (2001) et Yang
et al.(2002). Mais les données obtenues sont traitées de manière
très variées.
b) Régression linéaire
Les rapports, doublement normalisés (au niveau des fluorochromes et
au niveau de l'expérience et de son témoin), servent au calcul
du rapport E/T de l'expression d'un gène i dans l'expérience
(E) comparé à son témoin (T).
A ce rapport d'expression, est associé un facteur de pondération.
Il sert à indiquer si un gène doit être pris en compte
ou non dans les données d'expression différencielle finales.
Il est basé sur la reproductibilité ou non des intensités
des signaux entre les deux fluorochromes (![]() i est inversé si ratio >1):
i est inversé si ratio >1):
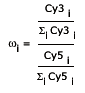 avec Cy.i : intensité du signal Cy. avant normalisation pour le gène
i
avec Cy.i : intensité du signal Cy. avant normalisation pour le gène
i
Ainsi deux intensités relatives identiques pour Cy3 et Cy5 donnent un
facteur de pondération à 1. Au contraire, deux intensités
relatives très différentes pour Cy3 et Cy5 entraînent un
facteur de pondération proche de 0. L'évaluation spot par spot
du facteur de pondération se fait sans a priori sur les intensités
du signal. Et il s'agit de l'indicateur final de la validation du rapport d'expression
du gène.
L'analyse des données s'appuie sur des hypothèses concernant des similitudes de signaux attendus entre les deux fluorochromes et entre l'expérience et son témoin. Tout d'abord, nous supposons que pour l'ensemble des gènes de l'expérience, le rapport des mesures d'expression en Cy3 sur Cy5 doit être égale à 1. De même, pour l'ensemble des gènes du témoin, le rapport des mesures d'expression en Cy3 sur Cy5 doit être égale à 1. Dans le laboratoire, la normalisation est basée sur une approche d'analyse de régression linéaire (Ugolin, 2002). Pour des échantillons très proches, la plupart des gènes sont attendus avec une expression à des niveaux constants. Un nuage de points des mesures des intensités de Cy5 en fonction de Cy3 doit avoir une pente de 1. De même, les intensités mesurées pour les témoins ajoutés en concentrations équimolaires doivent avoir un comportement similaire. Sous cette hypothèse, on peut calculer la pente de la droite de régression. Elle est utilisée pour ajuster les données par rotation afin d'obtenir une pente de 1. Soit ai le rapport corrigé de signal Cy3 sur le signal Cy5 pour le gène i.
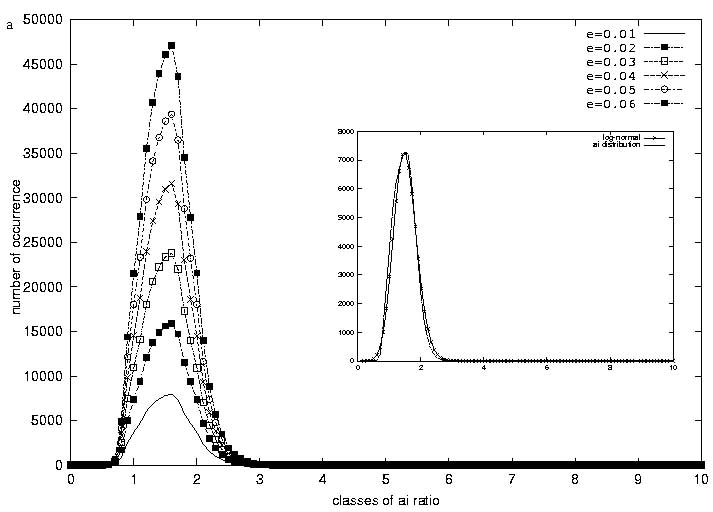 |
Fig.10 Dénombrement des ratios d'expressions expérience/témoin. |
Une dernière
hypothèse est que le ratio des expressions expérience/témoin
doit être égale à 1 dans la population des gènes
invariants. L'ensemble des rapports ai / aj (pour tous gènes i,j avec
i différent de j) tel que le ratio a=E/T, est calculé (E = expérience
et T = témoin).
Ces ratios sont attendus proche de 1 pour des gènes modulés de
la même manière. On dénombre la quantité de rapports
ai / aj contenus dans une fenêtre [1-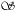 ,
1+]. Les ratios forment alors une même classe de niveau d'expression.
Le calcul est répété pour
toutes les valeurs de ? comprises entre 0,01 et 0,06 (Fig. 10). La classe la
plus importante comprend la population des gènes invariants, et ai indique
le rapport sur lequel elle est centrée. Les ratios ai sont recentrés
autour de 1 en divisant par la valeur a de la population des gènes invariants
ainsi déterminée. Par régression linéaire, la pente
du nuage de ratios est corrigé en fonction de cette population de gènes
invariants. Localement la population de gènes invariants peut décrocher
de la droite de régression. Pour palier cette irrégularité,
le nuage de points est centré sur la droite de régression à l'aide
d'une fenêtre glissante, qui calcule pour chaque position le décalage
entre la moyenne de la population stable, délimitée par les bornes
de la fenêtre, et la position de la droite de régression correspondant
au centre de la fenêtre. Les points délimités par les bornes
de la fenêtre sont corrigés par translation perpendiculairement à la
pente de la droite de régression. Ainsi la population stable a des ratios
ai égale à 1 pour E/T (Fig. 11).
,
1+]. Les ratios forment alors une même classe de niveau d'expression.
Le calcul est répété pour
toutes les valeurs de ? comprises entre 0,01 et 0,06 (Fig. 10). La classe la
plus importante comprend la population des gènes invariants, et ai indique
le rapport sur lequel elle est centrée. Les ratios ai sont recentrés
autour de 1 en divisant par la valeur a de la population des gènes invariants
ainsi déterminée. Par régression linéaire, la pente
du nuage de ratios est corrigé en fonction de cette population de gènes
invariants. Localement la population de gènes invariants peut décrocher
de la droite de régression. Pour palier cette irrégularité,
le nuage de points est centré sur la droite de régression à l'aide
d'une fenêtre glissante, qui calcule pour chaque position le décalage
entre la moyenne de la population stable, délimitée par les bornes
de la fenêtre, et la position de la droite de régression correspondant
au centre de la fenêtre. Les points délimités par les bornes
de la fenêtre sont corrigés par translation perpendiculairement à la
pente de la droite de régression. Ainsi la population stable a des ratios
ai égale à 1 pour E/T (Fig. 11).
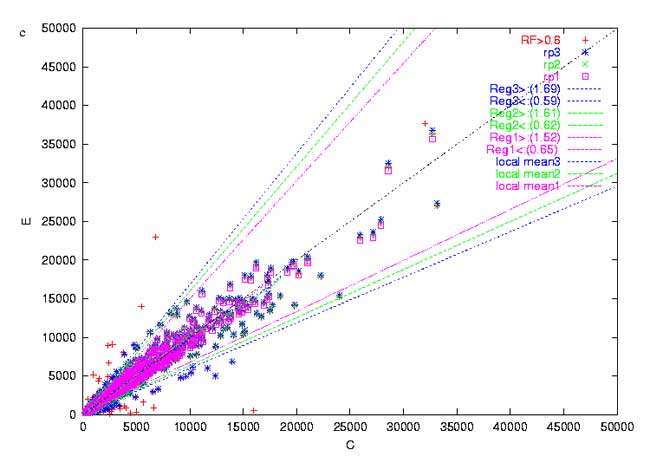 |
Fig.11 Normalisation des ratios d'expression expérience/témoin |
II. Logiciels d'analyse d'image pour le traitement
des données
A. Positionnement automatique des grilles de lecture
1. Recherche des objets présents dans l'image
Le but du positionnement automatique des grilles de lecture est d'isoler
les spots sur la lame. La difficulté est d'arriver à distinguer les
spots des traces diverses (poussières, traces de doigts,...). L'efficacité du
traitement de l'image pour éliminer le bruit de fond global sur la lame
déterminera la qualité de l'identification des spots. Les filtres
habituellement utilisés pour améliorer l'image sont des filtres
de type passe-bas. Ils sélectionnent l'intensité d'un pixel suivant
celles de 8 pixels environnant. Ces filtres ont la particularité de
lisser l'image. En contre partie, ils propagent les erreurs au niveau du pixel
voisin. Ceci n'a pas d'importance pour localiser le spot, mais ne convient
pas à la quantification du signal. Pour éviter cela, le laboratoire
a développé ses propres filtres pour d'une part délimiter
la position des spots, et d'autre part, pour quantifier la fluorescence. La
localisation du spot nécessite l'identification précise du bruit
de fond et des signaux parasites. Et le filtre idéal à cette étape
doit pouvoir supprimer tout le bruit de fond en dehors du spot (Fig.
12).
 |
Fig.12a Image
de la puce avant le passage du filtre
|
 |
Fig.12b
Bruit de fond qui est soustrait à l'image de la puce à ADN.
|
2. Détermination de la zone d'analyse (spot)
La grille des spots est calculée à l'aide de la fréquence
du dépôt , par des méthodes faisant appel à la transformée
de Fourier (). Le seul préalable reste uniquement le nombre de blocs
en lignes et en colonnes sur la puce à ADN. Cette donnée est
fournie dans le descriptif de la puce délivré par l'automate.
Le programme détermine automatiquement le nombre, la position et la
taille des spots. L'image du scanner ne commençant pas toujours aux
mêmes coordonnées, les coordonnées des spots, également
fournies par l'automate, ne peuvent pas être utilisées.
B. Repositionnement manuel par interface graphique
Bien que la majorité des grilles de lecture des spots soient
correctement positionnées, certaines lames très bruitées
présentent des grilles avec le mauvais nombre de lignes ou de
colonnes. Parfois un bloc entier de spots n'est pas identifié ou
une grille a été placée sur une zone couverte
d'artefacts. Un contrôle visuel reste indispensable pour vérifier
le positionnement des grilles. Et parfois une intervention manuelle
est nécessaire. Plutôt qu'une correction fastidieuse via
un fichier répertoriant toutes les modifications à apporter,
le développement d'une interface graphique permet d'effectuer
les ajustements par de simples clics de souris. En plus de l'affichage
de l'image de la lame et des caractéristiques des spots détectés
automatiquement, l'interface doit pouvoir répondre aux actions
de l'utilisateur. Cette interaction avec l'utilisateur est commune à toutes
les interfaces. Un outil de développement, comme Qt Designer,
fournit toute une bibliothèque d'objets et un protocole
original pour solliciter et capter ces interactions.
1. Environnement de développement Qt Designer
a) Librairie d'objets C++
La classe de base de la librairie de Qt est la classe QWidget. Le widget
est l'atome central de l'interface avec l'utilisateur: il reçoit
les événements produits à la souris et au clavier
et il se représente lui-même à l'écran.
Un widget qui n'est pas inclus dans un widget parent est dit au premier
niveau. Généralement un widget de premier niveau correspond à la
fenêtre de l'application avec un cadre et une barre de titre.
Un widget qui n'est pas de premier niveau, est un widget «enfant».
Il est inclus dans un widget parent, et souvent il ne se distingue
pas visuellement du parent. Par exemple, un bouton est un type de widget,
qui sera inséré dans un autre widget, tel qu'une fenêtre
de dialogue.
La classe QWidget est dérivée en un ensemble de sous-classes,
comportant des fonctionnalités directement utilisables, telles
que les classes QPushButton (bouton), QPopupMenu (menu surgissant),...
Le widget a une fonction appelée en réponse à un événement.
Il peut réagir à un clic de souris en réimplémentant
la fonction mousePressEvent().
b) Particularités de Qt Designer pour interagir avec l'utilisateur
Qt dispose d'un mécanisme de communication entre objets, appelé signal/slot.
Dans la programmation d'interface graphique, on souhaite souvent qu'un
changement sur un widget soit notifié à un autre widget.
Les anciens kits d'outils utilisent des «callbacks». Il
s'agit d'un pointeur sur une fonction. Si vous souhaitez qu'une fonction
active vous informe d'un événement, vous passez un pointeur
sur une autre fonction (le callback) à la fonction active. La
fonction active appelle ensuite le callback au moment approprié.
Les callbacks présentent deux inconvénients majeurs:
la fonction active peut appeler le callback avec des arguments de mauvais
type, et le callback est fortement couplé à la fonction
active, car la fonction active doit connaître quel callback
appeler.
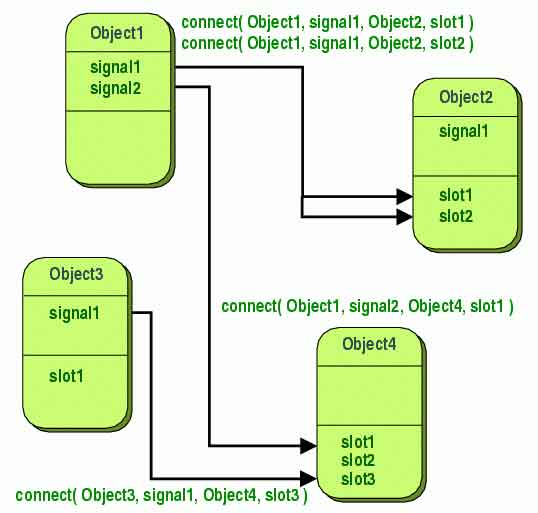 |
Fig.13 Représentation schématique
des connections entre signaux et slots
|
L'alternative proposée par Qt est l'utilisation des signaux et des slots (Fig. 13). Un signal est émis quand un événement particulier se produit. Les widgets ont plusieurs signaux et slots pré-définis, mais il est toujours possible d'ajouter ses propres signaux et slots. A un signal, doit correspondre au moins un slot de réception (contrôle fait par le compilateur). Toutefois le signal n'a pas à connaître quel slot le recevra. Et un slot est une fonction membre normale, qui ne sait pas quel signal lui est connecté. Le slot sera appelé avec les paramètres du signal au bon moment, avec autant d'arguments de n'importe quel type souhaités. L'information est réellement encapsulée.
2. Objectifs de
l'interface Surfview
L'interface doit permettre le repositionnement des grilles de lecture des spots
sur l'image de la puce à ADN. L'application doit donc prendre en charge
l'affichage de l'image au format TIFF produite par le scanner. Ensuite elle
reprend les informations sur les spots obtenues par la détection automatique.
Ces données servent à la représentation des spots sur
l'image en arrière-plan. La plupart des actions concerne un bloc de
spots désigné par l'utilisateur: modification du nombre de lignes
et de colonnes, ajout ou suppression du bloc complet, déformation du
bloc, maximisation ou ajustement de la taille des spots du bloc. Ces actions
sont interprétées par le logiciel en fonction du contexte. De
manière plus ponctuelle, l'utilisateur pourra souhaiter changer la taille
et la position d'un spot en particulier. Lorsque toutes les modifications ont été apportées,
les nouvelles caractéristiques des spots doivent être enregistrées
dans un fichier de même nom que le fichier généré par
la détection automatique, au quel est ajouté «_T» pour
préciser qu'il a été transformé, ou simplement
contrôlé si la pose automatique a réussi parfaitement.
3. Modélisation et conception de l'interface
Le point de départ de notre modèle est une classe appelée
ScribbleWindows, dérivée de QWidget. Elle permet de définir
les fonctions essentielles : un cadre affiché à l'écran
comportant en haut un menu «Fichier» pour accéder à l'image
de la lame et aux caractéristiques des spots détectés
automatiquement, et pour enregistrer les nouvelles caractéristiques
des spots. A côté du menu «Fichier», le menu «Options» sert à modifier
des paramètres d'affichage, et le menu «Aide» rappelle le
but du programme et les raccourcis clavier pour les utilisateurs expérimentés.
Chaque élément des menus appelle une fonction membre de la classe
ScribbleWindow via un slot. Ainsi lorsqu'un choix est signalé par l'utilisateur,
le programme peut lancer l'action adaptée définie dans la fonction.
La visualisation d'une image en haute définition nécessite souvent l'affichage d'une partie de l'image à l'écran. Le reste de l'image est accessible via des ascenseurs qui permettent de décaler la zone visible. Qt fournit la classe QScrollView pour définir ces ascenseurs. Une nouvelle instance de cet objet est déclarée dans la classe ScribbleWindow. Elle peut recevoir en argument le nom du widget qui sera pris en charge par les ascenseurs. En l'occurrence, ce sera une instance de la classe ScribbleArea dans laquelle sera chargée l'image et les représentations des spots. Donc ScribbleWindow possède deux «enfants»: la barre de menus et l'objet QScrollView, qui se charge lui-même de ScribbleArea.
Une surcharge de la méthode virtuelle resizeEvent() sert à réattribuer les bonnes dimensions aux ascenseurs lorsque l'utilisateur modifie la taille d'affichage de l'application. De plus, elle informe l'instance de ScribbleArea du changement de taille, en lançant un signal (reloadShow). Ce signal est connecté à la fonction de ScribbleArea en charge du réaffichage des spots (slotReloadShow). Ainsi l'information d'une modification au niveau de la classe parent peut être transmise à la classe enfant.
L'essentiel des
fonctions du programme est en fait géré au niveau
de la classe enfant, ScribbleArea. Les différentes fonctions de cette
classe sont les suivantes:
1- Constructeur: initialisation de variables de classe, composition du menu
popup, raccourcis clavier.
2- Destructeur: libération de la mémoire pour les variables créées
par allocation dynamique.
3- Ouverture de l'image de la puce à ADN, placée en arrière
plan
4- Ouverture du fichier *.sfr contenant les coordonnées initiales des
spots
5- Classement des blocs, avec des numéros croissants en allant de gauche à droite, et de haut en bas
6- Enregistrement du fichier *_T.sfr pour sauvegarder les nouvelles coordonnées
des spots
7- Recopie des caractéristiques d'un spot dans un autre
8- Transfert de ligne et de colonne de spots d'un bloc
9- Suppression de ligne et de colonne de spots d'un bloc
10- Suppression de bloc
11- Ajout de ligne et de colonne de spots d'un bloc
12- Déformer le bloc
13- Recherche de la position d'un bloc par rapport à ses voisins
14- Recherche de la position d'un spot par rapport à ses voisins
15- Distance entre le centre du spot et son centre relatif dans l'espace
délimite
par ses voisins
16- Recherche du chevauchement entre blocs.
17- Recherche du chevauchement entre spots. Réduction du diamètre
si chevauchement.
18- Connexité pour faire apparaître un objet dans l'image
entoure par un spot.
19- Connexité sur l'ensemble du bloc
. Slot pour recevoir l'instruction de l'utilisateur
. Réduction de la taille des spots ne contenant pas d'objet
. Connexité des spots du bloc dans l'ordre des densités décroissantes
20- Transfert d'un spot sélectionné dans le cas de la déformation
de bloc
21- Changement du diamètre d'un spot sélectionné
22- Affichage des spots
23- Gestion des options d'affichage
24- Interactions avec les événements de la souris
Toutes ces fonctions sont transparentes pour l'utilisateur. Il dispose d'un menu d'actions sur un bloc. Pour sélectionner le bloc, il presse le bouton droit de la souris à l'intérieur du bloc. Il apparaît un menu surgissant (ou popup), qui lui propose des actions sur une rangée de spots ou sur le bloc entier. Lorsqu'une action est choisie par un clic, le menu disparaît et l'action est exécutée. Si l'utilisateur passe par le raccourcis clavier, le menu reste affiché, et une autre action est possible sur le bloc toujours sélectionné. Ainsi après un court temps de prise en main, les chercheurs peuvent enchaîner la suppression de plusieurs colonnes, puis rajouter une ligne et immédiatement optimiser la taille des spots du bloc.
La dernière classe d'objet de ce projet est la classe qui définit un spot. Elle est enfant de la classe ScribbleArea. Lorsqu'un spot est créé, il reçoit les caractéristiques déterminées lors de la détection automatique: coordonnées sur la puce, diamètre, distance entre voisins à la verticale et à l'horizontale, son numéro de ligne et de colonne dans le bloc. De plus, il peut contenir les dimensions et les coordonnées d'un rectangle délimité par ses voisins, le nombre de pixels allumés dans le spot, des pointeurs sur le spot précédent et le spot suivant, et l'indication s'il est en cours de sélection ou non. Une fonction (spotShow) permet au spot de se représenter lui-même à l'écran. Elle intègre une classe QPainter, qui sert à dessiner des objets graphiques. La fonction drawChord est utilisée pour tracer un arc de 360°. Ainsi le cercle obtenu laisse toujours l'image de la lame visible en arrière plan. Il est coloré en rouge par défaut, et en vert s'il est en cours de sélection.
4. Implémentation
de l'interface
L'affichage d'image et de dessins nécessite des ressources mémoires
importantes. Les spots disposent d'une classe objet relativement simple, qui
leur permet de se représenter eux-même à l'écran.
Etant dérivée d'une classe QWidget, elle hérite de la
possibilité d'être intégrée dans un widget parent.
Lors de l'instanciation d'un nouveau spot, il reçoit en argument ScribbleArea,
désigné par «this». Dans le constructeur du spot,
ce paramètre permet de préciser à l'objet Spot qu'il est
intégré dans ce widget parent.
inline Spot::Spot(QWidget* widget = 0) { scribble = widget; ...};
La déclaration est inline pour accélérer la création
des milliers de spots de la puce à ADN.
L'objet spot se superpose à l'image chargée en arrière-plan.
Comme il est transparent par défaut, l'image est toujours visible. Le
spot apparaît lorsque la fonction showSpot() est appelée.
inline void Spot::spotShow()
{
QPainter p(scribble);
if(SELECT) p.setPen(QColor(0x00, 0xFF, 0x00));
else p.setPen(QColor(0xFF, 0x00, 0x00));
p.drawChord((int)(xs-dias/2),(int)(ys-dias/2),dias,dias,0,16*360);
}
Elle crée alors un objet QPainter capable de dessiner dans ScribbleArea,
qui est passé en argument de classe (scribble). Suivant que le spot
est en cours ou non de sélection, il est représenté avec
une couleur rouge ou verte. Le cercle est obtenu par une fonction qui dessine
un arc sur 360°. Ainsi le milieu du cercle reste transparent, avec l'image
en arrière plan.
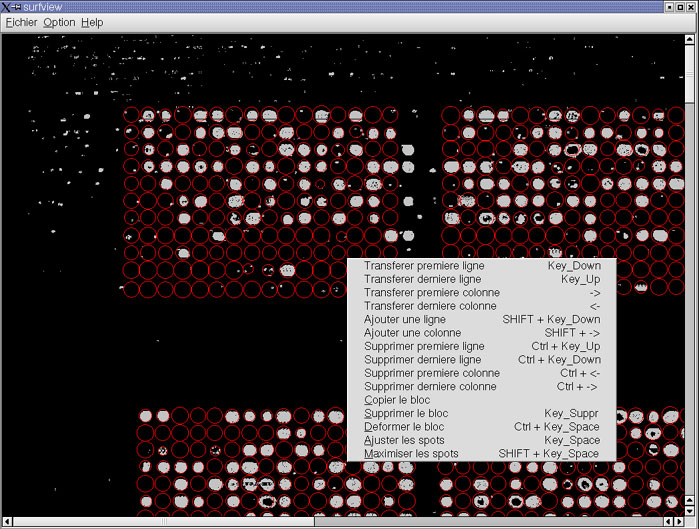 |
Fig.14
Interface Surfview après ouverture d'une image de puce à ADN
et création des spots obtenus Pour
accélérer les corrections, les modifications
peuvent être appelées par des raccourcis clavier. |
La création des spots est lancée par l'utilisateur lorsqu'il choisit le fichier contenant les caractéristiques des spots obtenues automatiquement (Fig. 14). La fonction Blocs de ScribbleArea reçoit le chemin pour ouvrir ce fichier. Elle crée dynamiquement la variable Bloc de dimension suffisante pour recevoir toutes les instances de Spot. Il s'agit donc d'un tableau à trois dimensions : Bloc[i][j][k], avec i le numéro de bloc, j le numéro de ligne et k le numéro de colonne du spot. Chaque Bloc[i][j][k] mémorise les informations sur un spot : coordonnées x et y sur l'image, diamètre, la distance horizontale au spot suivant (lambda x), la distance verticale au spot suivant (lambda y), les valeurs des indices j et k. Par ailleurs, le diamètre moyen des spots est calculé en additionnant les diamètres de tous les spots créés, et en divisant la somme par leur nombre total. Le nombre de lignes et de colonnes du bloc i sont mémorisés dans les variables plan_row[i] et plan_col[i], respectivement. Si le fichier contient bien des données sur les spots, la variable booléenne SPOTCREE devient TRUE. Elle sert principalement à autoriser l'utilisateur d'agir sur les spots uniquement lorsqu'ils sont créés.
Une fois les spots créés, l'ordre des blocs est vérifié pour que les numéros de bloc (indice i) soient croissants pour les blocs répartis de gauche à droite, et de haut en bas sur l'image de la puce à ADN. Pour cela, un tableau intermédiaire capable de pointer sur les adresses des blocs permet de classer les blocs, en les ordonnant les uns après les autres suivant les positions relatives du premier spot du bloc. Ensuite quatre variables enregistrent quels sont les voisins en haut, en bas, à droite et à gauche d'un bloc donné.
Par la suite, les appels de fonctions dépendent des modifications que les chercheurs souhaitent apporter aux positions des spots. Avec un clic gauche, l'utilisateur peut désigner un spot donné. Avec un clic droit, il peut sélectionner un bloc donné.
Lorsqu'un spot est sélectionné, la fonction transfert_spot() est exécutée. Cette dernière détermine en fonction de la position de la souris quel est le spot «visé». La variable SELECT du spot devient TRUE, et sa couleur change alors du rouge au vert. La variable DEPLACE passe également en TRUE. Elle sert à distinguer les fonctions à appeler suivant les actions à la souris. Le diamètre du spot peut alors être augmenté ou diminué de 1 pixel par appel des fonctions slot_aug_dia() ou slot_dim_dia() via les raccourcis claviers (Page_Up ou Page_Down). Si l'utilisateur déplace la souris , le spot est déplacé tant qu'il reste encadrer par ses voisins. Le contrôle de l'encadrement par les voisins se fait dans la fonction mouseMoveEvent(). Un nouveau clic de souris «redépose» le spot avec ses nouvelles caractéristiques. SELECT et DEPLACE repassent en FALSE. Une nouvelle action peut alors être lancée.
Lorsqu'un bloc est sélectionné, un menu d'actions à effectuer sur le bloc apparaît. Le placement automatique des grilles déterminant le nombre de lignes et de colonnes du bloc peut être trompé par la présence de poussières sur la lame. Si tous les blocs sont généralement trouvés, le nombre de rangs de spots doit parfois être corrigé. L'utilisateur choisit donc dans le menu s'il veut ajouter, supprimer ou transférer une ligne ou une colonne. Chaque option est rattachée à une fonction appelée par un signal. Le bloc sélectionné est déterminé en fonction de la position de la souris au moment de l'appel du menu, qui est comprise ou non entre les quatre spots aux «coins» du bloc. Dans le cas d'un ajout, la mémoire suffisante est réallouée pour contenir une nouvelle rangée de spots. L'écartement moyen entre les spots sert au décalage de la nouvelle rangée de spots par rapport à l'ancien bord du bloc, et au calcul du diamètre des nouveaux spots. Dans le cas d'une suppression, les instances de spot inutile sont supprimées. Les indices des spots doivent être réarrangés pour supprimer uniquement l'indice de ligne ou de colonne le plus élevé. Sinon les tableaux de Bloc risqueraient de contenir des valeurs NULL à certains indices. Pour les transferts, il s'agit d'une recopie des caractéristiques des spots dans les bonnes cases du tableau Bloc. Le décalage du rang à transférer est déterminé par un écartement moyen comme dans le cas d'ajout d'un rang.
Le bloc entier peut aussi être supprimé, copié ou déformé. La suppression de bloc consiste à renuméroter les blocs. Le bloc supprimé se retrouve à la fin du tableau Bloc[b]. Comme le nombre de blocs (memobloc) est diminué de 1, le bloc supprimé n'est plus pris en compte dans le programme. Le destructeur le détruira définitivement lorsque l'application est quittée. La variable memobloc_total lui indique le nombre réel de blocs créés. Le classement des blocs et les nouvelles «relations de voisinage» sont déterminés après avoir supprimer le bloc. Ainsi lorsqu'un bloc est copié, il faut tenir compte des éventuels blocs à «recycler» avant de créer de nouvelles instances de spots. Si le nombre de rangées dans le bloc recyclé est insuffisant, il faut réallouer de la mémoire pour ces rangées supplémentaires. A l'inverse, les colonnes et les lignes en trop sont supprimées. Si aucun bloc n'est recyclable, un nouveau bloc est formé avec de nouvelles instances de Spot. Dans cet espace mémoire ajustée aux bonnes dimensions, les caractéristiques du bloc qui sert de modèle sont recopié. Les coordonnées x, y du premier spot du bloc copié sont mémorisées. Le premier spot du nouveau bloc passe alors en mode sélectionné. Il peut être déplacé pour indiquer le nouvel emplacement du bloc. La variable BLOCOPIE passe en TRUE pour répondre de manière adaptée aux actions à la souris. Lors du déplacement de la souris, le spot sélectionné en vert suit le curseur tant que le bloc dispose d'une place suffisante pour être copié. Le spot sélectionné retourne à sa position initiale si le nouveau bloc devait chevaucher les blocs voisins. Le nouvel emplacement est choisi par un clic de souris. La translation appliquée au premier spot est répétée aux autres spots du bloc. Si le premier spot est resté à sa position initiale, le bloc est supprimé suivant la méthode précédemment. Le classement et les relations entre blocs sont réactualisés.
La déformation de bloc répond à une attente particulière des chercheurs. Les spots ont normalement un alignement horizontale et verticale très homogène grâce à la dépose automatique. Toutefois certains «glissements» de blocs apparaissent occasionnellement sur les lames. Les blocs se retrouvent alors avec une orientation transversale, qui présente toujours une certaine régularité. La solution apportée par le programme est simple et précise. Le chercheur peut placer les quatre coins d'un bloc à l'emplacement qu'il désire. Un repositionnement fin est toujours possible au cours de la procédure. Une fenêtre de dialogue demande la confirmation de la transformation lorsque les quatre coins du bloc ont été placés. Les positions des autres spots sont recalculées pour se placer de manière régulière entre ces quatre pôles. Le même diamètre, le plus grand possible sans qu'il n'y ait de chevauchement entre spots, est attribué à tous les spots.
 |
Fig.15 Maximisation
de la taille
des spots Tout le signal sera pris en compte lors de l'analyse. |
 |
Fig.16 Ajustement
de la taille
des spots La majorité des signaux est identifiée et isolée au plus juste. Certains signaux sont toutefois légèrement tronqués. Dans la pratique, il sera privilégié la méthode de maximisation. |
Les deux dernières options du menu concernent la zone qui sera utilisée pour l'analyse du signal. Comme la méthode d'analyse du signal permet d'identifier le bruit de fond intra-spot et inter-spot, elle possède une grande fiabilité. Il importe donc de définir une zone d'analyse qui inclut tout le signal, même si le diamètre du spot est légèrement surestimé. Toute la difficulté est d'isoler un seul dépôt sur la lame, et surtout d'éviter d'exclure une partie du signal. Ainsi l'option «Maximiser les spots» permet d'attribuer le plus grand diamètre possible à chacun des spots du bloc sélectionné, sans qu'il n'y ait de chevauchement avec les spots voisins (Fig. 15). Pour privilégier la taille de la zone d'analyse des signaux les plus forts, une chaîne sert à classer les spots du bloc dans l'ordre des densités décroissantes. La densité correspond au rapport du nombre de pixels identifiés comme un objet sur le diamètre du spot. Au final tous les spots sont contigus avec leur voisin. Mais le diamètre des spots ne contenant pas de signal se retrouve suffisamment réduit pour laisser la place nécessaire aux spots contenant les signaux. L'option «Ajuster les spots» reprend la même procédure, mais réduit la taille du spot au plus prêt de l'objet détecté sur l'image (Fig. 16). L'objet est recherché par connexité dans une zone elliptique délimitée par les voisins du spot. Dans la pratique, il est préférable de privilégier la maximisation qui prend la totalité du signal.
Avec les signaux correctement identifiés, la mesure de l'intensité peut être normalisée suivant la méthode décrite dans la partie I.B. Les probabilités d'expression des gènes peuvent alors être déterminées.
C. Probabilités
de chaque état d'expression associées à un
gène
1. Modulation
d'expression d'un gène: invariabilité, induction et
répression
L'induction et la répression sont les termes utilisés pour se
référer à l'expression des gènes dans des cellules
d'une expérience comparée à un témoin. Un gène
induit a une expression plus forte dans les cellules de l'expérience
que dans les cellules du témoin. A l'inverse, un gène réprimé a
une expression plus faible dans les cellules de l'expérience que dans
celles du témoin. Cela peut être formulé également
par: un gène réprimé a une expression plus forte dans
les cellules du témoin que dans celles de l'expérience. Il
est à noter que la définition de la répression prend
en compte cette possibilité d'inverser le témoin et l'expérience,
car le terme désigne une comparaison purement relative.
L'expression du gène est invariable lorsqu'elle est au même niveau dans les cellules de l'expérience et du témoin. Il peut donc s'agir d'une expression très forte du gène, qui donnera un signal fluorescent élevé proche entre l'expérience et le témoin. Mais le gène invariant peut aussi avoir une expression très faible voire nulle dans les cellules de l'expérience et du témoin.
2. Symétrisation des rapports d'expression
Lorsque le rapport d'expression E/T est calculé, l'intensité du
signal de l'expérience est divisée par celle du signal du témoin.
A l'égalité d'expression entre les deux lots de cellules, ce
rapport est égale à 1. Dans le cas d'une très forte
expression pour l'expérience et d'une expression quasi nulle pour
le témoin,
ce rapport tend vers l'infini. A l'inverse, si l'expression est très
forte pour le témoin et quasi nulle pour l'expérience, le rapport
tend vers 0. Les intervalles [1,+![]() [ et ]0,1] apparaissent clairement dissymétriques.
Pour rétablir la symétrie, la majorité des publications
proposent de calculer le log2 du rapport d'expression E/T. Ainsi un gène
exprimé deux fois plus dans l'expérience comparé au
témoin,
a un rapport qui augmente d'une unité de log. Et un gène exprimé deux
fois plus dans le témoin comparé à l'expérience,
a un rapport qui diminue d'une unité de log. La symétrie semble
donc rétablie pour une distribution gaussienne d'un ensemble de gènes.
Toutefois l'échelle d'observation étant tassée pour
les répressions, la variabilité des mesures a un impact beaucoup
plus important pour les gènes réprimés que pour les
gènes
induits (Tran, 2002). En d'autres termes, la moindre erreur sur la répression
se retrouve très nettement amplifiée. De plus, la distribution
des rapports ne suivent pas une loi pure. Il s'agit plutôt d'un mélange
de différentes lois pour les différentes classes d'induction.
Dès lors la transformation par le log ne garantit plus la conservation
de la symétrie entre les gammes d'induction et de répression.
[ et ]0,1] apparaissent clairement dissymétriques.
Pour rétablir la symétrie, la majorité des publications
proposent de calculer le log2 du rapport d'expression E/T. Ainsi un gène
exprimé deux fois plus dans l'expérience comparé au
témoin,
a un rapport qui augmente d'une unité de log. Et un gène exprimé deux
fois plus dans le témoin comparé à l'expérience,
a un rapport qui diminue d'une unité de log. La symétrie semble
donc rétablie pour une distribution gaussienne d'un ensemble de gènes.
Toutefois l'échelle d'observation étant tassée pour
les répressions, la variabilité des mesures a un impact beaucoup
plus important pour les gènes réprimés que pour les
gènes
induits (Tran, 2002). En d'autres termes, la moindre erreur sur la répression
se retrouve très nettement amplifiée. De plus, la distribution
des rapports ne suivent pas une loi pure. Il s'agit plutôt d'un mélange
de différentes lois pour les différentes classes d'induction.
Dès lors la transformation par le log ne garantit plus la conservation
de la symétrie entre les gammes d'induction et de répression.
En se fondant sur la définition des rapports d'induction et de la répression, il apparaît possible de[...me contacter pour cette partie...]
Un autre point consiste maintenant à distinguer les gènes invariants des gènes induits. Pour cela, nous supposons l'existence de deux distributions différentes entre les gènes induits et les gènes réprimés.
3. Distribution gaussienne des gènes invariants
Notre analyse repose sur deux hypothèses:
- les gènes invariants suivent une distribution normale, lorsqu'un échantillon
est hybridé contre lui même plusieurs fois (Brody, 2002).
- l'ensemble
des gènes induits constitue une succession de distributions gaussiennes
se chevauchant sur [0, +![]() [.
[.
En effet, en reproduisant au laboratoire un très grand nombre de fois
une expérience, les facteurs d'induction mesurés pour tous
les gènes induits suivent des distributions gaussiennes se chevauchant.
En s'appuyant sur ces deux critères, nous allons cherché,
d'une part, à estimer la distribution des gènes invariants à l'aide
d'une distribution normale, et d'autre part, à trouver une loi de
distribution permettant de refléter la résultante du chevauchement
de toutes les classes d'induction sur [0, +![]() [.
Un même traitement
est effectué sur ]-
[.
Un même traitement
est effectué sur ]-![]() , 0[. Il permet d'obtenir la gamme de répression.
, 0[. Il permet d'obtenir la gamme de répression.
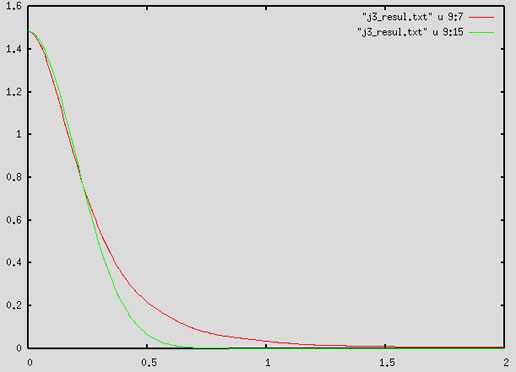 |
Fig.17
Distribution réelle et distribution de la loi normale --- distribution de la population réelle --- distribution théorique des gènes invariants |
Pour trouver les
lois décrivant au mieux les lois de distribution des
gènes invariants, nous avons procédé de la manière
suivante: [...me
contacter pour cette partie...]
4. Distribution
log-normale modifiée des gènes induits
Inversement aux gènes invariants, si une population des gènes
induits existe dans la population réelle, elle devient majoritaire
vers les modulations élevées. [...me
contacter pour cette partie...]
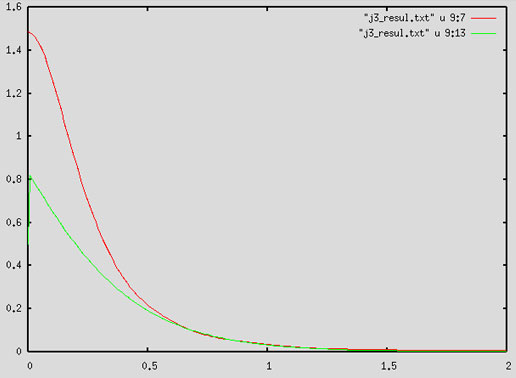 |
Fig.18
Distribution réelle et distribution de la loi log-normale
modifiée --- distribution de la population réelle --- distribution théorique des gènes induits (loi log-normale) |
5. Mélange de lois pour représenter l'ensemble de l'expression
des gènes
[...me contacter pour cette partie...]
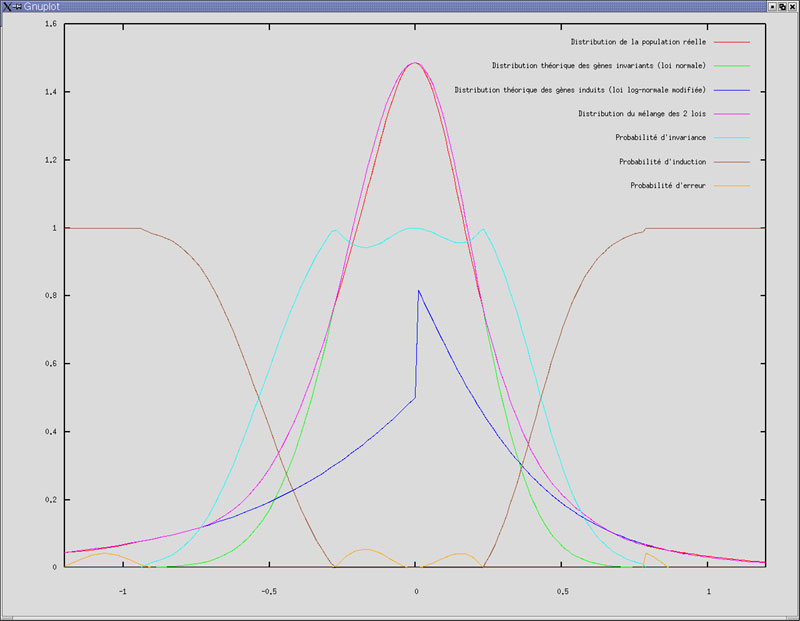 |
Fig.19
Représentation des distributions réelle et théoriques,
ainsi que les probabilités d'expression Nous
constatons la dissymétrie des courbes théoriques
d'induction. En effet, le nombre de gènes induits n'a pas de |
Ceci permet de déterminer la probabilité d'induction ou d'invariance de l'expression d'un gène (Fig. 19). Le même calcul est réalisé pour la partie symétrique des gammes d'induction. Ainsi les probabilités d'invariance, d'induction ou de répression peuvent être établies.
Au cours de cette étude, nous avons montré les techniques d'obtention des données de puces à ADN et leur influence sur le traitement de l'analyse d'image. Le volume d'informations nécessite un traitement automatique dont le processus est en cours de développement. Au niveau de la pose des grilles de lecture des signaux, nous avons fourni un nouvel outil au laboratoire LCE du CEA. Maintenant les chercheurs contrôlent et éventuellement corrigent les zones analysées sur les puces à ADN, grâce à une interface graphique conviviale et fiable.
Pour la validation des données, les répétitions limitées pour un gène particulier ont été compensées par un protocole expérimentale de reverse labeling, avec une correction et une normalisation adaptées. Grâce au facteur de pondération retranscrivant la reproductibilité des mesures et à la segmentation du signal distinguant le bruit de fond intra- et inter-spot, des modulations d'expression d'un gène entre une expérience et son témoin peuvent être établies de manière fiable. La symétrisation originale de ces modulations s'appuie sur le caractère relatif de la différence d'expression entre une expérience et son témoin. La recherche d'une loi gaussienne décrivant la distribution des gènes invariants et d'une loi log-normale modifiée décrivant la distribution des gènes induits peut alors s'effectuer successivement sur les domaines d'induction et de répression. Les probabilités d'induction, d'invariance et de répression de l'expression d'un gène sont déduites des deux lois obtenues sur chacun des domaines.
Les niveaux d'expression simultanés de milliers de gènes à différents temps durant le processus biologique permettent de regrouper les gènes suivant ces profils temporels de niveau d'expression. La classification des gènes par reconnaissance de leurs profils d'expression s'inscrit par exemple dans le cadre de recherche d'activité de molécules pharmaceutiques ou de diagnostique clinique. La suite de cette étude pourra consister à intégrer les probabilités d'expression du gène dans les techniques de classification.
Sigles
ADN: Acide désoxyribonucléique. Macromolécule formée
de désoxyribonucléotides, qui constitue le matériel génétique
de toutes les cellules eucaryotes, des cellules procaryotes et de certains
virus.
ADNc: ADN complémentaire. ADN simple brin, qui est une copie d'un ARN
obtenue par une transcription inverse. L'ADNc double brin résulte de
la copie du premier brin par une ADN polymérase.
ADN polymérase:
Enzyme catalysant la polymérisation (5' vers
3') des mononucléotides triphosphates qui constituent l'ADN.
ARN: Acide ribonucléique. Macromolécule formée de ribonucléotides
résultant de la transcription de l'ADN, présente dans les cellules
eucaryotes, les cellules procaryotes, ainsi que dans certains virus.
ARNm: Acide ribonucléique messager. ARN qui va être traduit en
séquence protéique.
EST: Expressed Sequence Tag
CEA: Commissariat à l'Energie Atomique.
LCE: Laboratoire de Cancérologie Expérimentale
NCBI: National Center for Biotechnology
Information.
PBS: Phosphate Buffer Saline. Tampon phosphate.
PCR: Polymerase chain reaction. Procédé d'amplification exponentielle
in vitro d'une séquence définie d'ADN, faisant intervenir des cycles
successifs d'appariements d'oligonucléotides spécifiques et d'élongation à l'aide
d'une polymérase.
SAGE: Serial Analysis of Gene Expression
TE: tampon Tris-EDTA
TIGR: The Institut for Genomic Research.
TNF: Tumor Necrosis Factor. Facteur de nécrose de tumeur.
Notes
Spot
homogène:
La zone, sur laquelle l'hybridation de molécule marquée est
régulièrement répartie, émet des signaux fluorescents
d'intensités proches sur l'ensemble de sa surface.
Unité d'hybridation:
Ensemble de molécules identiques déposées sur une surface
délimitant un spot.
Sigma
réel: Un
moyen d'optimisé la recherche de la loi théorique consiste à prendre
comme Sigma réel, la largeur de la distribution réelle à mi-hauteur
au pic principal, centré sur zéro, car cette grandeur se rapproche
de l'écart-type de la distribution des gènes invariants.
RMS: Le RMS est non cumulé, car le nombre de valeurs entre les différents
tests est constante.
Bibliographie
Brody
James, et al., 2002 – Significance
and statistical errors in the analysis of DNA microarray data, PNAS, Vol.
99, 20:12975-12978
Diehl Frank, et al., 2001 – Manufacturing DNA microarrays of high spot
homogeneity and reduced background signal, Nucleic Acids Research, Vol. 29,
No. 7 e38.
Dixon Arthur et Damaskinos Savvas, 2001 – Confocal scanning of genetic
microarrays, DNA Arrays: methods and protocols, Humana Press, Methods in Molecular
Biology, Vol.170, 16:237-246
Hegde Priti, et al., 2000 – A concis guide to cDNA microarray analysis
, Biotechniques 2000 Sep;29(3):548-50, 552-4, 556 passim
International Human Genome Sequencing Consortium, 2001 – Initial sequencing
and analysis of the human genome, Nature, 409:860-921.
Kerr K.M. et Churchill G.A., 2001 – Experimental design for gene expression
microarrays, Biostatistics, 2:183-201.
Martinez M. Juanita et al., 2003 – Identification and removal of contaminating
fluorescence from commercial and in-house printed DNA microarrays, Nucleic
Acids Research, Vol. 31, No. 4 e18
Stoecklin Georg et al., 2003 - A Constitutive Decay Element Promotes Tumor
Necrosis Factor Alpha mRNA Degradation via an AU-Rich Element-Independent Pathway,
Molecular and Cellular Biology, May 2003, Vol. 23, No. 10: 3506-3515
Tran Peter et al., 2002 - Microarray optimizations: increasing spot accuracy
and automated identification of true microarray signals, Nucleic Acids Research,
Vol. 30, No. 12 e54.
Ugolin Nicolas et al., 2002 – Brevet US n° 10/173.672 du 19/06/2002,
CA n° 2389901 du 20/06/2002.
Ugolin Nicolas et al., 2002 – A highly effective experimental design
and data processing method to improve the discovery potential of cDNA microarray
analysis (en presse).
Ugolin Nicolas et al., 2003 – Instrumental and statistical approach for
image analysis from microarrays (bientôt soumis).
Velculescu V. et al., 1997 – Serial Analysis of Gene Expression, Science,
270:484-487.
Venter J. Craig et al., 2001 - The Sequence of the Human Genome, Science 2001
February 16, 291: 1304-1351
Yang Y. Hwa et al., 2002 – Normalization for cDNA microarray data: a
robust composite method addressing single and multiple slide systematic variation,
Nucleic Acids Research, Vol. 30, No. 4 e15, 10 p.
![]()